Evolutionary Genome Mining
Last updated on 2026-02-20 | Edit this page
Overview
Questions
- What is Evolutionary Genome Mining?
- Which kind of BGCs can EvoMining find?
- What do I need in order to run an evolutionary genome mining analysis?
Objectives
- Understand EvoMining pipeline.
- Run an example of evolutionary analysis in cpsG gene family.
- Explore MicroReact interactive output interface.
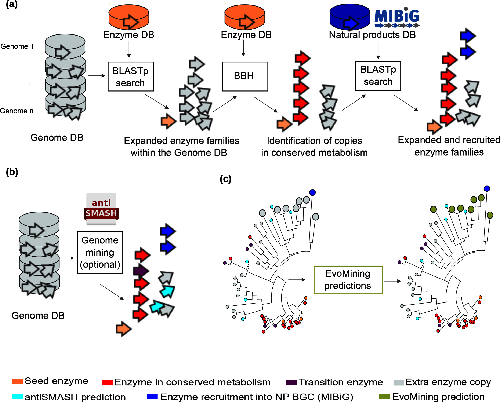
Usually, bioinformatics tools related to the prediction of Natural Products (NP) biosynthetic genes try to find metabolic pathways of enzymes that are known to be related with the synthesis of secondary metabolites. However, these approaches fail for the discovery of novel biosynthetic systems. Thus, EvoMining tries to circumvent this problem by detecting novel enzymes that may be implicated in the synthesis of new natural products in Bacteria.
To know more about EvoMining you can read Selem et al, Microbial Genomics 2019.
EvoMining searches protein expansions that may have evolved from the conserved metabolism into a specialized metabolism. It builds phylogenetic trees based on all the protein copies of a certain enzyme in a given genome database. The output tree differentiates copies that are related with the conserved metabolism, copies that are known to be implicated in discovered NP-producing-BGCs i.e. BGCs from MiBIG database and, optionally, protein copies that belong to BGCs predicted by antiSMASH. Finally, some branch in the tree will be depicted as “EvoMining hits”, which represent enzyme expansions that are evolutionary closer to those copies related with the secondary metabolism (MiBIG or antiSMASH BGCs) than to those related with the conserved (primary) metabolism.
Run evomining image
First, place yourself at your working directory.
OUTPUT
CORASON_GENOMES Corason_Rast.IDs cpsg.query GENOMES outputThe general structure of a docker container is shown in the next
bash-box. Note that it requires to specify which docker container will
run. Optionally, with -v flag it is possible to share a
directory with the container, with -p flag a port is shared
and it is also possible to specify which program will run inside the
container.
BASH
$ docker run --rm -i -t -v <your local directory>:<inside docker directory> -p <inside port>:80 <docker container> <program inside docker> EvoMining is inside a docker container, so the general structure to start your analysis will be as follows:
BASH
$ docker run --rm -i -t -v $(pwd):/var/www/html/EvoMining/exchange -p 8080:80 nselem/evomining:latest /bin/bash Let’s explain the pieces of this line.
| command | Explanation |
|---|---|
| docker | tells the system that we are running a docker command |
| run | the command that we are running is to run a docker container |
| --rm | this container will be removed after closed |
| -i | this container allows user interaction |
| -t | this interaction will be through a terminal |
| -v | a data volume (directory) will be shared between your local machine and the container |
| -p | a port will allow a web based app |
However, sometimes the port 80 is busy, in that case you can use
other ports like 8080 or 8084. If this is the case, please use the port
80X where X is a number between 01..30 provided by your
instructor.
BASH
$ docker run --rm -i -t -v $(pwd):/var/www/html/EvoMining/exchange -p 8080:80 nselem/evomining:latest /bin/bash
$ docker run --rm -i -t -v $(pwd):/var/www/html/EvoMining/exchange -p 8084:80 nselem/evomining:latest /bin/bash If your docker container worked, now you will see in your terminal a
new prompt. Instead of the usual dollar sign, there should be a number
# at the beginning of your terminal. This is because now
you are inside the docker container and you have sudo
permissions inside the docker.
To exit container use exit
And now your prompt must be back in the dollar sign
Set EvoMining genomic database
Start the container again with your corresponding port.
BASH
$ docker run --rm -i -t -v $(pwd):/var/www/html/EvoMining/exchange -p 80X:80 nselem/evomining:latest /bin/bash Though we will NOT run the test EvoMining command, it looks as follows:
# perl startEvoMining.pl Instead of that, customize the genomic database by using the same as
CORASON.
Notice that EvoMining requires RAST-like annotated genomes and for this
reason we are using the fasta files that CORASON converts from our gbk
inputs.
# perl startEvoMining.pl -g GENOMES -r Corason_Rast.IDs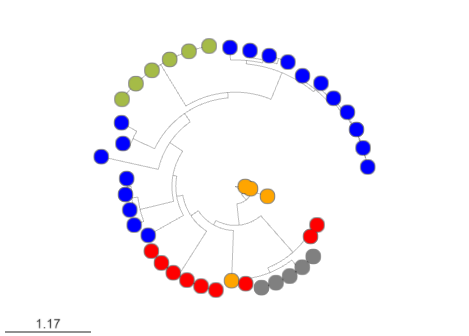{kind=link}

Finally, remember that X means your user-number and open
your browser at the address:
http://132.248.196.38:80X/EvoMining/html/index.html. Once
there, just click the start button and enjoy! (click on the submit
buttons!)
When you finish using this container, please exit it.
#exitVisualize your results in MicroReact
Firstly, you have to run all the pipeline in the website: http://
EvoMining outputs are stored in the directory
<conserved-db>_<natural-db>_<genomes-db>
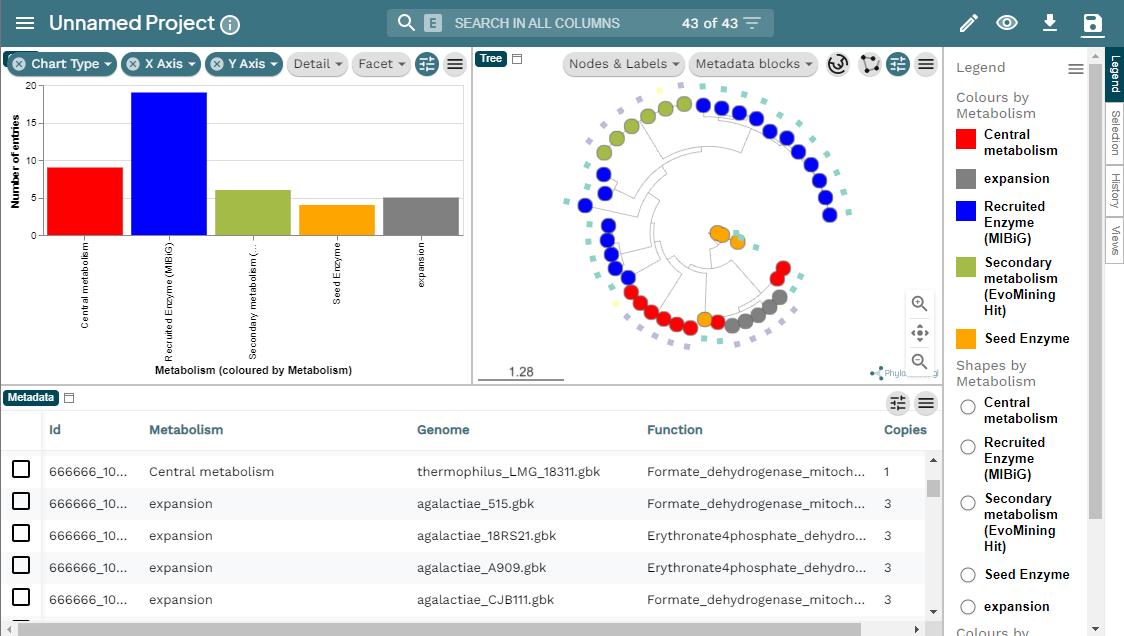
To explore EvoMining outputs, you need to upload 1.nwk and 1.csv files to microReact. There are many methods to download files from the server to your local computer.
If you are using JupyterHub you explore the file folders and select the files and then press the download button.
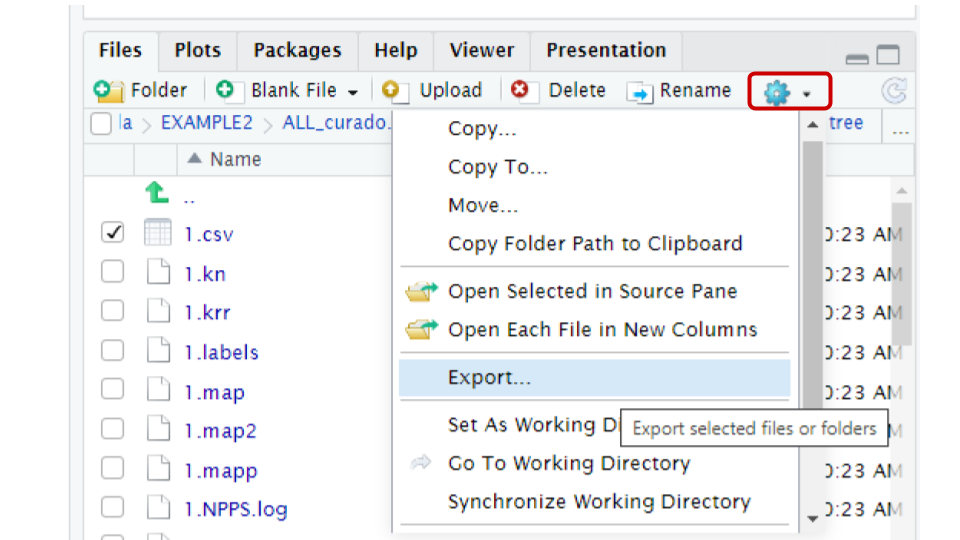
You can use the export button in the file panel of R studio. To
download the files, first in the files panel open in the directory
~/pan_workshop/results/genome-mining/corason-conda/EXAMPLE2/ALL_curado.fasta_MiBIG_DB.faa_GENOMES/blast/seqf/tree,

Then, select files 1.nwk and 1.csv in that directory, click
more in the engine icon, and select the export
option in the menu. The files will be downloaded to your local computer,
and now you will be able to upload them to MicroReact.
Alternatively, If your prefer to use the terminal to download files
the scp protocol can download the files into your local
machine.
BASH
scp betterlab@132.248.196.38:~/pan_workshop/results/genome-mining/corason-conda/EXAMPLE2/ALL_curado.fasta_MiBIG_DB.faa_GENOMES/blast/seqf/tree/1.nwk ~/Downloads/.
scp betterlab@132.248.196.38:~/pan_workshop/results/genome-mining/corason-conda/EXAMPLE2/ALL_curado.fasta_MiBIG_DB.faa_GENOMES/blast/seqf/tree/1.csv ~/Downloads Here you can find the MicroReact visualization of this EvoMining run.
Other resources
To run EvoMining with a larger conserved-metabolite DB you can use EvoMining Zenodo data.
To explore more EvoMining options, please explore EvoMining wiki.
Set the conserved-enzymes database
When using EvoMining, oftenly you will desire to construct your own conserved enzymes database. To know more about how to configure a database, consult the EvoMining wiki in the EvoMining databases part. Natural products database could also be replaced for another set of genes that are “true positives”, for example a set of regulatory genes.
As an example, transform the file cpsg.query into the
format of this database. This file contains the aminoacid sequence of
the cpsG gene. Firstly, copy this file into what will become
the conserved-enzymes database.
Now, it requires some editing. Open nano editor and change the first
line >cpsg to
>SYSTEM1|1|phosphomannomutase|Saga. EvoMining
conserved-database needs a four-field format pipe-separated that
contains; the name of the metabolic system to which the enzyme belongs
(SYSTEM1), a consecutive number of the enzyme (1 in this case), the
function of the enzyme, and finally, an abbreviation of the organism
Saga, (S. Agalactiae).
The reason behind this is that this was the way we needed EvoMining for
its first use and we have not changed the headers since.
OUTPUT
>SYSTEM1|1|phosphomannomutase|Saga
MIFVTVGTHEQQFNRLIKEVDRLKGTGAIDQEVFIQTGYSDFEPQNCQWSKFLSYDDMNSYMKEAEIVITHGGPATFMSVISLGKLPVVVPRRKQFGEHINDHQIQFLKKIAHLYPLAWIEDRun your EvoMining docker
BASH
$ docker run --rm -i -t -v $(pwd):/var/www/html/EvoMining/exchange -p 80X:80 nselem/evomining:latest /bin/bash and inside this new container:
# perl startEvoMining.pl -g GENOMES -r Corason_Rast.IDs -c cpsg_cdbUse the website again and think about the results.
Exercise 1. Set EvoMining parameters
Complete the blanks in the following EvoMining run:
actinoSMASH A file with the ids of antiSMASH recognized
genes. Actinos a directory with RAST-like fasta and
annotation files. Histidine-db A fasta file with some
proteins in the histidine pathway.Actinos.ids tabular files with the RAST ids and the name of
the organisms.
# perl starEvoMining.pl -g ____ -c _____ -r _____ -a ___________ Discussion 1. Retro EvoMining in enzyme database
What do you learn from running in a conserved-enzymes database the gene cpsG that is part of a specialized BGC?
cpsG does not have extra copies in Streptococcus agalactiae, so there are no expansions that may be functional divergent. cpsG single copies in the genomes look red-colored in EvoMining output, as if they belong to the conserved-metabolism. However, this is not the case, the color is because there is only one copy and it is merged into MIBiG true-positives because it was originally a gene in the specialized metabolism. So it is important to know the seed enzymes.
OUTPUT
cpsg_cdb_MiBIG_DB.faa_GENOMESARTS is another evolutionary genome mining software with its corresponding database ARTS-db .
Evolutionary Genome Mining
To learn more about Genome Mining you can read these references:
- The confluence of big data and evolutionary genome mining for the discovery of natural products. Chevrette et al, 2021`
- Evolutionary Genome Mining for the Discovery and Engineering of Natural Product Biosynthesis. Chevrette et al, 2022
More about docker
To see the running container use
ps
If there are containers in use you will see a list of all of them.
OUTPUT
2f879ba6e337 nselem/evomining:latest "/bin/bash" 11 hours ago Up 11 hours 0.0.0.0:8014->80/tcp, :::8014->80/tcp relaxed_dirac
- EvoMining is a command-line tool that performs evolutionary genome mining over gene families
- EvoMining hits can belong to new BGC
- MicroReact is an interactive genomic visualizer compatible with EvoMining output